产品








服务


联系我们
400-021-7882
021-54281913
021-54281913
|
mRNA疗法作为一种新兴的治疗方法,在遗传医学领域展现出巨大潜力,其应用范围涵盖癌症免疫疗法、蛋白替代疗法、基因治疗和疫苗开发。mRNA脂质纳米颗粒(LNPs)通过封装mRNA分子,实现其在体内的高效递送。然而,该领域的技术复杂性和工作流程的多样性为研究人员进入这一领域设置了障碍。本文旨在提供一个易于操作的mRNA LNP体外评估的详细指南,以降低研究门槛,促进学术界、工业界和临床环境中的研究进展。 |
|||||
01
|
|||||
|
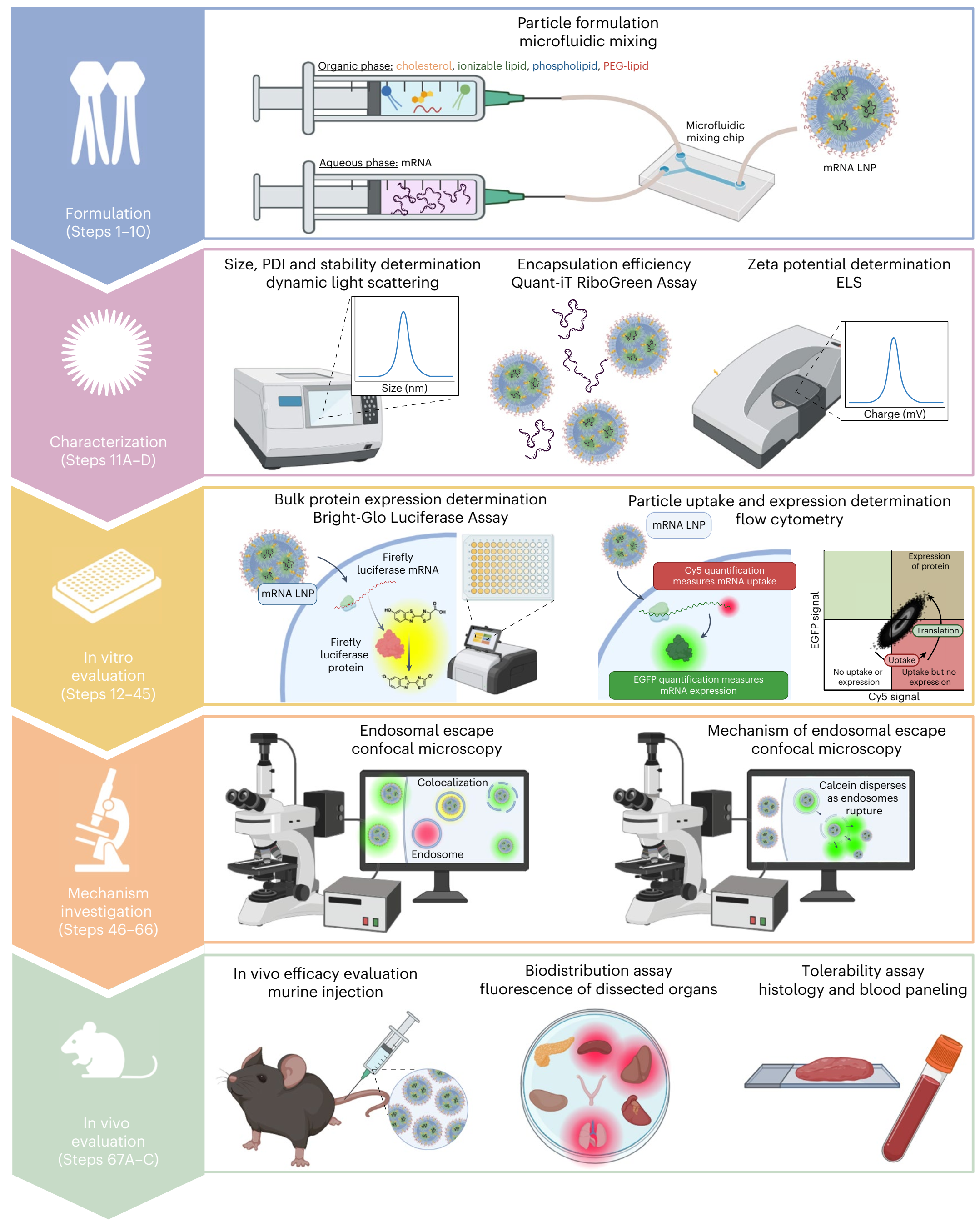
研究使用SM-102、DOPE、胆固醇和C14-PEG-2000,摩尔比为48:10:40:2为配方。通过微流控混合技术制备mRNA-LNP。为了证明方案可以应用于制备和评价各种mRNA LNP配方,实验还制备了添加不同辅料的mRNA LNPs。 通过流式细胞术定量评估LNP的体外蛋白表达和细胞摄取,使用报告基因(如荧光素酶(FLuc)和Cy5-EGFP(Cy5信号指示细胞内mRNA的存在,EGFP信号反映目标蛋白的表达)。还通过共聚焦显微镜定性可视化蛋白表达和细胞摄取。 通过共聚焦显微镜研究mRNA LNPs的机制,主要研究两个方面:细胞摄取后从内质网逃逸的LNPs比例以及驱动LNPs从内质网释放的机制。在本方案中,研究者提供了一种利用Atto-488标记的mRNA LNP(绿色)和Lysotracker Deep Red染色内质网(红色)来研究mRNA从内质网逃逸的方法。通过WCIF ImageJ软件使用Pearson相关系数(PCC)定量测量内质网逃逸能力。为了评估质子海绵效应的影响,我们使用了bafilomycin A1(质子海绵效应抑制剂)和calcein(膜不透性染料)。 |
|||||
|
图 1 :mRNA -LNP的配方制定及评估方案概述。 |
|||||
02
|
|||||
| 2.1 mRNA-LNP的体外评估方法: | |||||
|
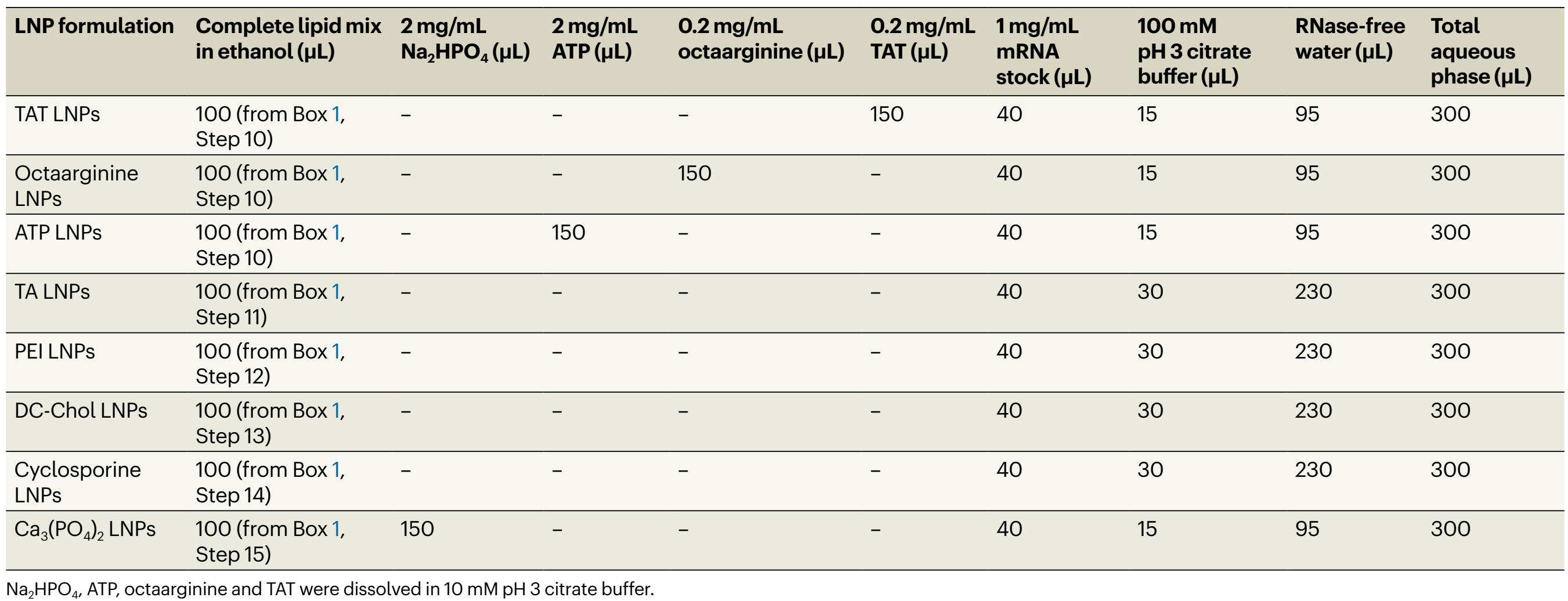
采用微流控制备的八种mRNA-LNP(表1)作为示例,评估细胞中FLuc表达、细胞活性和细胞摄取情况。使用FLuc mRNA-LNP,裸FLuc mRNA、新鲜培养基和Lipofectamine 2000进行细胞转染,以评估FLuc的细胞表达。使用1%(v/v)的Triton作为细胞活性测定的阴性对照。鉴于肝脏是许多mRNA脂质纳米颗粒配方的主要靶器官,选择HepG2细胞系(一种肝癌细胞系)作为示例。细胞使用 mRNA-LNP(每孔 100 μL,总 mRNA 浓度为 500 ng/ml,n = 3)处理 24 小时,然后使用 Bright-Glo 荧光素酶检测系统测量 FLuc 表达,并使用 alamarBlue 检测法测量细胞活性。 |
|||||
|
表 1:制备的 mRNA LNP 配方详情 |
|||||
|
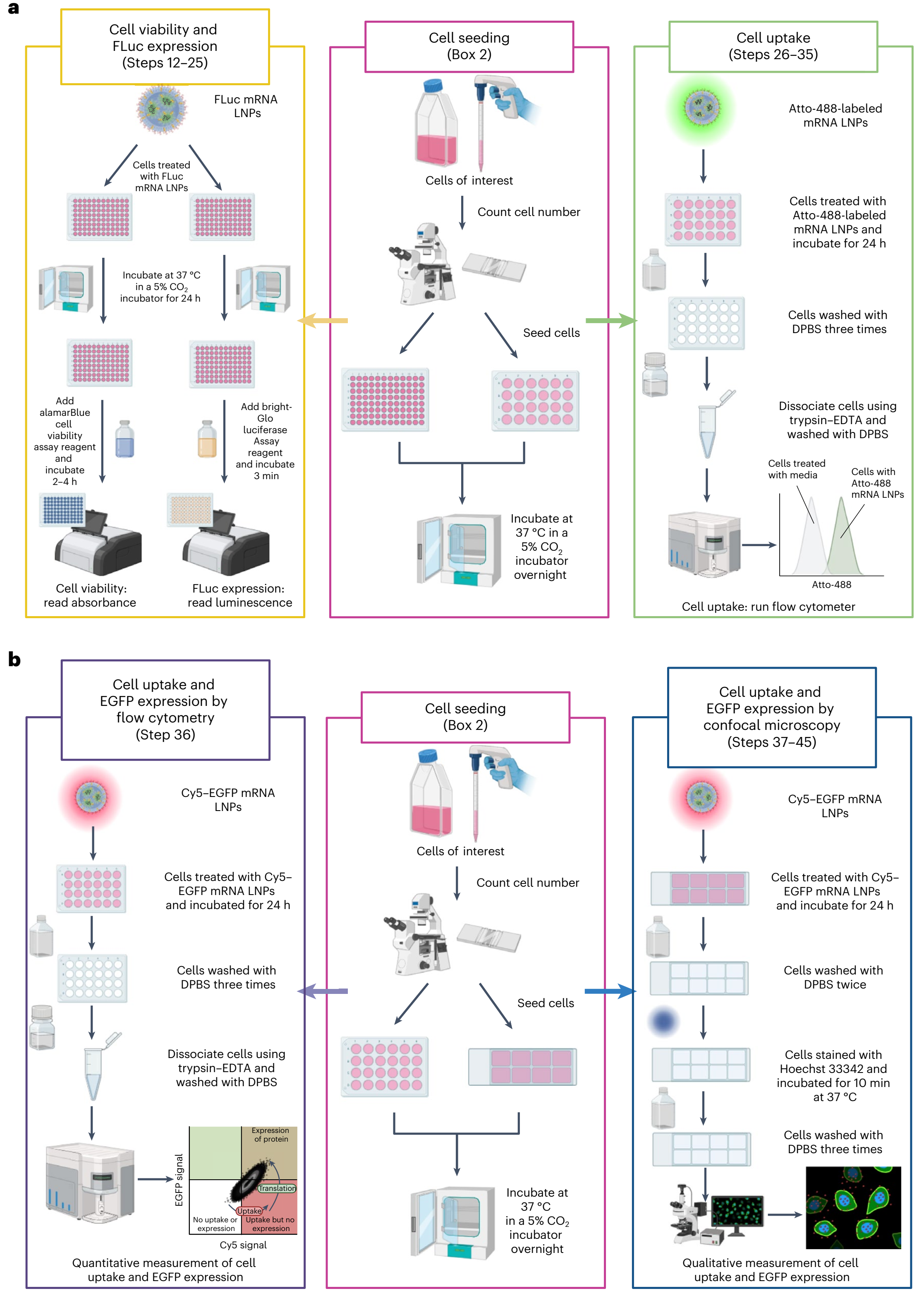
图2 :mRNA 脂质纳米颗粒体外评估方法概述 |
|||||
|
使用 Atto-488 标记的 DOPE制备mRNA LNP。随后将 Atto-488 标记的 mRNA-LNP 稀释至 500 ng/mL 的浓度。细胞用 Atto-488 标记的 mRNA LNP 进行处理(每孔 400 μL,总 mRNA 浓度为 500 ng/mL,n = 3)。用新鲜培养基处理的细胞作为对照。24 小时后,用不含酚红的胰蛋白酶-EDTA 洗涤并收集细胞,通过流式细胞术进行定量。 在本方案中,还采用了一种替代方法,利用 Cy5-EGFP(增强型绿色荧光蛋白)mRNA 同时定量蛋白质表达和细胞摄取。将 Cy5-EGFP mRNA LNP 稀释至 500 ng/mL 的浓度。细胞用 Cy5-EGFP mRNA-LNP(每孔总 mRNA 量为 400 μL 500 ng/mL,n = 3)处理。24 小时后,用不含酚红的胰蛋白酶-EDTA 洗涤并收集细胞。可根据具体应用需求调整孵育时间和细胞类型。Cy5 信号表明细胞内存在 mRNA,而 EGFP 信号反映目标蛋白的生成情况。两种荧光信号均可通过流式细胞术进行定量,四象限控制可提供有关以下细胞百分比的信息: 1.未摄取 mRNA(Cy5(-)/EGFP(-) 2.空白(即预期无细胞的象限)(Cy5(-)/EGFP(+) 3.已摄取了 mRNA 但尚未翻译(Cy5(+) / EGFP(-)) 4.这两种细胞都摄取了 mRNA 并将其翻译成了蛋白质(Cy5(+) / EGFP(+))。 此信息在图 2b 中有所展示。此外,还可以使用共聚焦显微镜进行可视化和定性定量分析。 |
|||||
| 2.2 机制研究方法: | |||||
|
通过将 HepG2 细胞与 Atto-488 标记的LNP共孵育,并使用共聚焦显微镜观察内体逃逸情况。用Atto-488 标记的 mRNA-LNP处理细胞。4 小时后,用溶酶体追踪试剂和 Hoechst 33342 对细胞进行染色。通过共聚焦激光扫描显微镜进行活细胞成像。在共聚焦图像中,细胞核呈蓝色,Atto-488 mRNA LNPs 呈绿色,内体呈红色,被内体捕获的 mRNA-LNP呈黄色(即绿色和红色信号的共定位)。此外,还可以使用 WCIF Image J 软件量化 PCC(该系数值为 0 表示 100%内体逃逸,值为 1 表示 0%内体逃逸),以评价 mRNA-LNP的内体逃逸能力。建议在 6 小时内研究 mRNA LNP 的溶酶体逃逸情况,因为通常 6 小时以下的孵育时间足以使 mRNA LNP 从溶酶体中逃逸出来。 为了研究质子海绵效应,采用了一种基于钙黄绿素染料的检测方法。细胞用新鲜培养基或含巴弗洛霉素 A1的培养基处理。然后将钙黄绿素加入这些细胞中,随后加入 LNP(LNP稀释至最终浓度为 500 ng/mL,n = 3)。使用未荧光标记的 mRNA 以避免干扰钙黄绿素信号。用钙黄绿素 + 巴弗洛霉素 A1 处理但不添加任何 mRNA 脂质纳米颗粒的细胞作为对照。4 小时后,通过共聚焦激光扫描显微镜进行活细胞成像。如果 mRNA 脂质纳米颗粒处理组中的钙黄绿素在整个细胞中扩散,这表明内体已破裂,mRNA 得以逃逸。如果 mRNA-LNP和巴弗洛霉素 A1 处理组中的钙黄绿素仅呈现小的点状分布,类似于细胞对照组,则表明质子海绵效应是其内体逃逸的潜在机制之一。建议孵育时间不要超过 4 小时,因为过长的孵育时间可能会降低巴弗洛霉素 A1 的抑制效果。 |
|||||
03
|
|||||
|
第 1 部分:mRNA LNPs 的体外效力评估 关键信息:根据应用的不同,可以选择不同的细胞系或细胞类型来评估 mRNA LNPs 的体外递送效力。使用前培养细胞并计数细胞数量。选择 HepG2 细胞系定量评估 mRNA LNPs 的细胞活性和体外荧光素酶表达 时间 2 - 3 天 1.向 96 孔板中加入 100 μL浓度为每 100 μL 1×10⁴ 个细胞的 HepG2 细胞。在 37°C、5%二氧化碳培养箱中孵育过夜,使细胞附着于培养板底。 2. 用 HepG2 细胞培养基(含 10%胎牛血清和 1%青霉素 - 链霉素的 DMEM 培养基)将 FLuc mRNA LNPs 稀释至最终浓度为 500 ng/mL。 3. 阳性对照包括裸mRNA 或 Lipofectamine 2000 与 mRNA 的复合物。 将 1 mg/ml的FLuc mRNA 用 HepG2 细胞培养基稀释至 500 ng/ml。制备Lipofectamine 2000 mRNA 对照:在 0.6 ml的试管中,加入 5μL Lipofectamine 2000 于 25 μL的 Opti-MEM 培养基中。在另一个 0.6 ml的试管中,加入 5 μL 1 mg/ml的裸露 FLuc mRNA 于 25 μL的 Opti-MEM 培养基中。将上述溶液混合,在室温下孵育 5 分钟。用 HepG2 细胞培养基将溶液稀释至最终浓度为 500 ng/ml。 4. 阴性细胞活性对照:在 HepG2 细胞培养基中稀释 Triton X-100 以获得 1%(体积比)的 Triton,作为阴性活性对照,以确保检测准确运行。 5.使用 2 ml的移液器将第1步中 96 孔板中的 HepG2 细胞培养基吸出并丢弃。 6.向三个孔(A4 - C4)中加入 100 μL 500 ng/ml的来自步骤2的FLuc mRNA LNP,向八个孔(A2 - H2)中加入 100 μL新鲜的 HepG2 细胞培养基(作为 100% 细胞活性对照),向三个孔(A6 - C6)中加入 100 μL 500 ng/ml的裸 FLuc mRNA,向三个孔(F12 - H12)中加入 100 μL 1%的 Triton。在 37°C、5% CO2 培养箱中孵育 24 小时。 7.进行 alamarBlue 细胞活性测定,在一个 15 ml的试管中,将 1 ml的 alamarBlue 细胞活性试剂加入到 9 ml的细胞培养基中(1:10)。 8.使用 2 ml的吸液管将第6步中 96 孔板上的上清液吸出并丢弃。 9.将步骤7中稀释的 alamarBlue 溶液倒入 25 ml的试剂储存器中。 10.使用多通道移液器向步骤8中制备的平板的每个孔中加入 100 μL的 alamarBlue 溶液。 11.将 96 孔板转移至 5%二氧化碳培养箱中,在 37°C 下孵育 2 至 4 小时。 12.在酶标仪中读取 570 纳米处的吸光度和 600 纳米处的背景值。 13.计算细胞活性 14.在另一块 96 孔板中,重复步骤1至6。向每个处理过的孔中加入 100 μL Bright-Glo 荧光素酶检测缓冲液。孵育 3 分钟,然后在微孔板读数仪上读取发光值。 |
|||||
|
第 2 部分:定量评估荧光标记LNP的细胞摄取情况 时间:2 - 3 天 关键信息:在 mRNA 脂质纳米颗粒配方中加入了 Atto-488 DOPE 分子(步骤 16)。 15.用 HepG2 细胞培养基将细胞悬液稀释至每 100 μL 2×10⁴ 个细胞的最终浓度。在 24 孔板的每个孔中加入 400 μL细胞。每个孔的最终细胞密度应为 8×10⁴ 个细胞。在 37°C、5% CO₂ 培养箱中孵育过夜,以使细胞附着于基质。 16.使用 Atto-488 DOPE 制备 Atto-488 标记的FLuc mRNA-LNP。用细胞培养基将 Atto-488 标记的 FLuc mRNA LNPs 稀释至最终浓度为 500 ng/mL。 17.用 2 ml的吸液管将 24 孔板中的上清液吸出并丢弃。 18.每孔向细胞中加入 400 μL 500 ng/ml的 Atto-488 标记的FLuc mRNA LNP。向其他细胞中加入 400 μL新鲜细胞培养基(作为未染色细胞对照)。在 37°C、5%二氧化碳培养箱中孵育 24 小时。 19.用 DPBS 洗涤细胞三次,每次向每个孔中加入 400 μL DPBS。孵育 10 秒,然后用2 ml吸液管吸取上清液并丢弃液体。 20.用 150 μL 2 倍浓度的胰蛋白酶 - EDTA(10 倍浓度胰蛋白酶 - EDTA 的 1:5 稀释液)使细胞分离,并在 5%二氧化碳培养箱中孵育 5 分钟。 21.一旦所有细胞从培养板上脱落,向每个孔中加入 200 μL DPBS,然后将每个孔中的细胞转移到单独的 1.7 ml管中。 22.用 DPBS 洗涤细胞两次,每次向每管中加入 400 μL DPBS,然后以 300g 离心 5 分钟,弃去上清液。每次洗涤后,将细胞悬浮于 200 μL DPBS 中。 23.在流式细胞仪(如 Attune NxT 流式细胞仪)上分析样本。将前向散射光(FSC)电压设为 100,侧向散射光(SSC)电压设为 280,并根据需要进行调整,以确保能清晰观察到单细胞簇。然后选择“BL1”通道来测量样本。 24.使用 FlowJo(或类似)软件,通过与未处理细胞对照相比具有更强荧光强度的细胞百分比或通过几何荧光平均强度来评估 Atto-488 标记的 FLuc mRNA LNPs 的摄取情况。 |
|||||
|
第 3 部分:使用荧光标记的LNP 的细胞摄取和蛋白质表达定量评估 时间 2 - 3 天 关键在信息:将 Cy5-EGFP mRNA 封装到所需的 LNP 配方中,该配方由可离子化脂质(SM-102)、磷脂(DOPE)、胆固醇和 PEG 脂质(C14-PEG-2000)以及 Cy5-EGFP mRNA 组成,制备方法参考上期内容。 25.在 24 孔板中,重复步骤15至22。使用流式细胞仪(如 Attune NxT 流式细胞仪)分析样本。将前向散射光(FSC)电压设为 100,侧向散射光(SSC)电压设为 280,并根据需要进行调整,以确保能清晰观察到单细胞簇。然后选择“BL1”通道和“RL1”通道来测量样本。使用 FlowJo 软件,通过将样本分为四个象限,以对照未处理细胞中荧光强度更强的细胞百分比来评估 Cy5-EGFP mRNA LNP 的摄取和表达情况。 26.计数 HepG2 细胞数量,然后用 HepG2 细胞培养基稀释细胞悬液,使最终浓度为每 100 μL 1×104 个细胞。向每孔加入 300 μL细胞,使最终细胞密度为每孔 3×104 个细胞(在 µ-slide 8 孔盖玻片载玻片中)。在 37°C、5% CO2 培养箱中孵育过夜,以使细胞附着于基质。 27.用细胞培养基稀释 Cy5-EGFP mRNA LNP,使其最终浓度为 500 ng/mL。 28.用 2 ml的抽吸移液管通过真空抽吸将 µ-slide 8 孔盖玻片载玻片上的上清液吸出并丢弃。 29.向每个孔中加入 300 μL 500 ng/ml的 Cy5-EGFP mRNA 脂质纳米颗粒。在 5%二氧化碳培养箱中 37℃孵育 24 小时。 30.用 DPBS 洗涤细胞两次,每次向每个孔中加入 300 μL DPBS。 31.在 15 ml的试管中,用 DPBS 将 10 mg/ml的 Hoechst 33342 稀释至 1 μg/ml。向每个孔中加入 300 μL 1μg/ml的 Hoechst 33342,然后在 37°C、5%二氧化碳培养箱中孵育 10 分钟。 32.重复步骤30,将细胞清洗三次。 33.向每个孔中加入 200 μL的 DPBS。 34.通过将 µ-slide 8 孔盖玻片置于共聚焦激光扫描显微镜下进行共聚焦显微镜活细胞成像,以观察 mRNA LNP 的水平(选择“Cy5”通道)和 EGFP 表达(选择“EGFP”通道)。使用 WCIF Image J 软件处理图像,添加比例尺并合并不同通道(蓝色代表细胞核,红色代表 Cy5-mRNA,绿色代表 EGFP 蛋白)。 |
|||||
|
第4部分:内体逃逸机制研究 时间 2 - 3 天 关键信息:为了量化细胞内 mRNA-LNP的内体逃逸能力,有必要在 LNPs 上添加一些荧光标记。使用 Atto-488 标记的 FLuc mRNA-LNP。 35.重复步骤 26。 36.重复步骤 16。 37.重复步骤 28。 38.向每个孔中加入 300 μL 500 ng/ml的 Atto-488 标记的荧光素酶 mRNA 脂质纳米颗粒。在 37°C、5%二氧化碳培养箱中孵育 4 小时。 39.重复步骤 30。 40.在 15 ml的试管中,用细胞培养基将溶酶体示踪剂深红色稀释至最终浓度为 100 nM。 关键步骤:将细胞培养基预热至 37 摄氏度。 41.向每个细胞孔中加入 300 μL 100 nM的稀释型溶酶体示踪剂深红色溶液,然后在 37 摄氏度、5%二氧化碳培养箱中孵育 1 小时。 42.重复步骤 30。 43.在 15 ml的试管中,用 DPBS 将 10 mg/ml的 Hoechst 33342 稀释至 1 μg/ml。向每个孔中加入 300 μL 1 μg/ml的 Hoechst 33342,然后在 37°C、5%二氧化碳培养箱中孵育 10 分钟。 44.重复步骤 30。 45.向每个孔中加入 200 μL的 DPBS。 46.通过将 µ-slide 8 孔盖玻片置于共聚焦激光扫描显微镜下进行活细胞成像,以可视化内体(选择“Lysotracker Deep Red”通道)、细胞核(选择“Hoechst 33342”通道)和 Atto-488 标记的 FLuc mRNA LNPs(选择“Atto-488”通道)。需要拍摄五张具有代表性的细胞图像(每张图像包含超过 20 个细胞)来计算 PCC 值。这些图像可以通过 WCIF Image J 软件进一步处理,以添加比例尺并合并不同通道(蓝色:细胞核;红色:内体;绿色:Atto-488 标记的 FLuc mRNA LNPs)。要获取 PCC 值,请将共聚焦图像导入带有“JACoP”插件的 WCIF ImageJ 软件。导航至“插件”,选择“JACoP”,然后选择“皮尔逊相关系数”。选择图像 A(绿色通道)和图像 B(红色通道),然后点击“分析”。软件将计算 PCC 值。内体逃逸能力通过 PCC 进行量化,其中 PCC 值为 1 表示无内体逃逸,而 PCC 值为 0 表示完全内体逃逸。 |
|||||
|
第5部分:使用巴弗洛霉素 A1 评估质子海绵机制 时间 1.5 天 47.重复步骤35至37,但在步骤36中,按照实验步骤制备未标记荧光素的 FLuc mRNA LNP。 48.向三个孔中加入 180 μL新鲜的 HepG2 细胞培养基,向另外三个孔中加入含有巴弗洛霉素 A1(111.1 nM)的新鲜 HepG2 细胞培养基。 49.在 1.7 ml的试管中,称取 1.5 mg的钙黄绿素,然后加入 1 ml的磷酸盐缓冲盐水(DPBS),使钙黄绿素的最终浓度为 1.5 mg/ml。 50.向每个孔中加入 20 μL 1.5 mg/ml的钙黄绿素,以获得 150 μg/ml的钙黄绿素和 100 nM的巴弗洛霉素 A1 的最终浓度。 51.向每个孔中加入 100 μL 1500 ng/ml的 mRNA 脂质纳米颗粒(以获得最终浓度为 500 ng/ml的 mRNA 脂质纳米颗粒)。向对照细胞中加入 100 μL新鲜培养基。在 37°C、5%二氧化碳培养箱中孵育 4 小时。 52.在 4 小时的孵育期结束后,用户可选择用 CellMask™ Deep Red Actin 跟踪染料(方案 A)对细胞骨架进行染色,或用 Hoechst 33342(方案 B)对细胞核进行染色,以分别观察细胞骨架和细胞核。 (A)染色细胞骨架 (i)在 15 ml的试管中,用细胞培养基将 CellMask™ Deep Red Actin 跟踪染料(1 毫摩尔)稀释至最终浓度为 1 微摩尔。 (ii)向每个处理过的孔中加入 300 μL 1 微摩尔的稀释 CellMask™ Deep Red Actin 跟踪染料。 (iii)在 5% CO2 培养箱中 37°C 孵育 15 分钟。 (B)染色细胞核 (i)用 Hoechst 33342 染色细胞核,请重复步骤 54。 53. 用 DPBS 洗涤细胞四次,重复步骤30。 54. 向每个孔中加入 200 μL的 DPBS。 55. 通过将 µ-slide 8 孔盖玻片置于共聚焦激光扫描显微镜下进行活细胞成像,以共聚焦显微镜观察钙黄绿素信号(选择“钙黄绿素”通道)、细胞核(选择“Hoechst 33342”通道)和细胞骨架(选择“CellMask Deep Red”通道)。图像可通过 WCIF Image J 软件进一步处理,以添加比例尺或合并不同通道(蓝色:细胞核;红色:细胞骨架;绿色:钙黄绿素)。 |
|||||
04
|
|||||
|
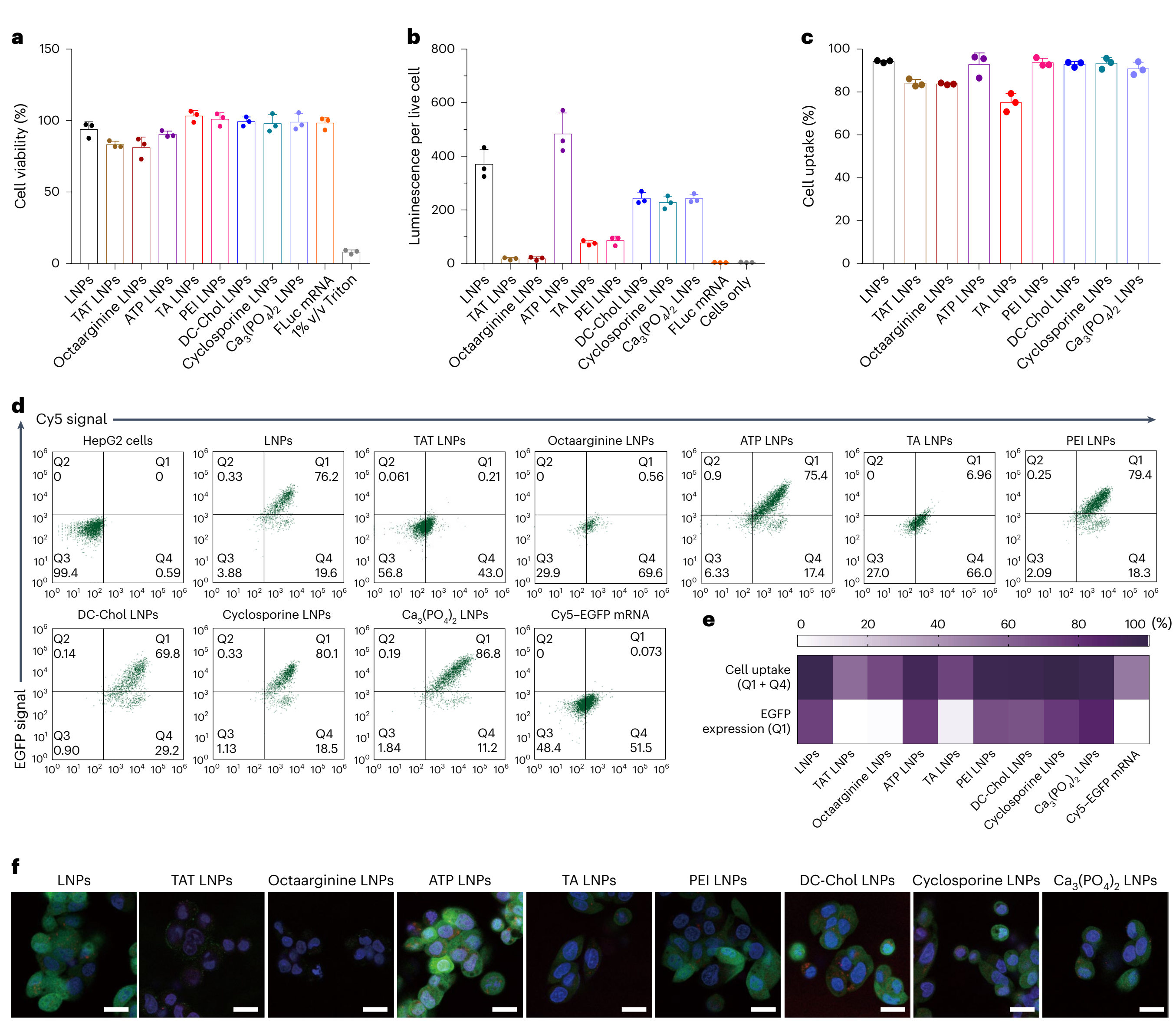
在HepG2 细胞的 FLuc 表达评估中,所有 mRNA LNPs 的耐受性均如预期般良好(图3a),1%的 Triton X-100 使细胞存活率低于 10%,这表明 alamarBlue 细胞活力测定法有效。在蛋白质表达方面,ATP LNPs 的 FLuc 表达量高于其他配方,而 TAT LNPs 和八精氨酸 LNPs 的 FLuc 表达量较低(图3b)。为了评估细胞摄取情况,利用 Atto-488 标记的 DOPE制备LNP ,并量化与培养基处理对照组相比荧光信号增强的细胞百分比。TAT LNPs、八精氨酸 LNPs 和 TA LNPs 的细胞摄取量低于 LNP 组(图3c)。将 Cy5-EGFP mRNA 封装到 LNP ,蛋白质表达和细胞摄取可通过流式细胞术进行定量评估(图3d、e),所得结果与图3b、c 中的结果相似。通过这种方法,可以同时评估蛋白质表达和细胞摄取。为了定性评估蛋白质表达和细胞摄取,可使用共聚焦显微镜可视化 Cy5-EGFP mRNA LNPs,为研究人员提供更直观、直接的 mRNA LNPs 效果评估方式。如图3f 所示,ATP LNPs 表现出更高的蛋白质表达,而 TAT LNPs、八精氨酸 LNPs 和 TA LNPs 则表现出较低的蛋白质表达,这与图3d、e 中的结果一致。 |
|||||
|
图3:mRNA-LNP的体外评估方法。 |
|||||
|
共聚焦显微镜图像表明 mRNA LNPs 被困在内体中(图4a)。TAT LNPs、ATP LNPs、TA LNPs、环孢素 LNPs 和 Ca3(PO4)2 LNPs 显示出更好的内体逃逸能力,这从较低的PCC值和代表性共聚焦显微镜图像中可以看出(图4b、c)。为了进一步评估内体逃逸的一个潜在机制——“质子海绵效应”,使用了质子海绵效应抑制剂(巴弗洛霉素 A1)和钙黄绿素(图4d)。加入巴弗洛霉素 A1 和钙黄绿素后,观察到只有在八精氨酸 LNPs 存在的情况下,钙黄绿素才会在整个细胞中扩散(图4e)。这表明“质子海绵效应”并非其内体逃逸的主要机制。相比之下,“质子海绵效应”似乎是所有其他 mRNA LNP 组内体逃逸的主要机制之一。 |
|||||
|
图4:机制研究方法 |
|||||
| 参考文献:Ma Y, VanKeulen-Miller R, Fenton OS. mRNA lipid nanoparticle formulation, characterization and evaluation. Nat Protoc. 2025 Mar 11. doi: 10.1038/s41596-024-01134-4. Epub ahead of print. PMID: 40069324. | |||||